An introduction to the RESOLVE R
package
Luca De Sano
Daniele Ramazzotti
April 07, 2026
Source:vignettes/RESOLVE.rmd
RESOLVE.rmdCancer is a genetic disease caused by somatic mutations in genes controlling key biological functions such as cellular growth and division. Such mutations may arise both through cell-intrinsic and exogenous processes, generating characteristic mutational patterns over the genome named mutational signatures. The study of mutational signatures have become a standard component of modern genomics studies, since it can reveal which (environmental and endogenous) mutagenic processes are active in a tumor, and may highlight markers for therapeutic response.
Mutational signatures computational analyses fall mostly within two categories: (i) de novo signatures extraction and (ii) signatures exposure estimation. In the first case, the presence of mutational processes is first assessed from the data, signatures are identified and extracted and finally assigned to samples. This task is typically performed by Non-Negative Matrix Factorization (NMF). While other approaches have been proposed, NMF-based methods are by far the most used. The estimation of signatures exposures is performed by holding a set of signatures fixed (see, e.g., COSMIC mutational signatures catalogue) and assigning them to samples by minimizing, e.g., mean squared error between observed and estimated mutational patterns for each sample.
However, available mutational signatures computational tools presents many pitfalls. First, the task of determining the number of signatures is very complex and depends on heuristics. Second, several signatures have no clear etiology, casting doubt on them being computational artifacts rather than due to mutagenic processes. Last, approaches for signatures assignment are greatly influenced by the set of signatures used for the analysis. To overcome these limitations, we developed RESOLVE (Robust EStimation Of mutationaL signatures Via rEgularization), a framework that allows the efficient extraction and assignment of mutational signatures.
The RESOLVE R package implements a new de novo signatures extraction algorithm, which employs a regularized Non-Negative Matrix Factorization procedure. The method incorporates a background signature during the inference step and adopts elastic net regression to reduce the impact of overfitting. The estimation of the optimal number of signatures is performed by bi-cross-validation. Furthermore, the package also provide a procedure for confidence estimation of signatures activities in samples.
Additionally, RESOLVE enables the precise stratification of cancer genomes based on mutational signatures, allowing for the identification of key signatures that drive specific cancer subtypes. Beyond de novo extraction, assignment, and confidence estimation, RESOLVE also supports patient clustering, survival analysis to identify risk groups linked to distinct mutational processes, and the investigation of associations between mutational signatures and driver gene mutations. These advanced capabilities offer deeper insights into tumorigenesis mechanisms, their prognostic relevance, and potential therapeutic targets.
As such, RESOLVE represents an addition to other Bioconductor packages, such as, e.g., SparseSignatures, MutationalPatterns, musicatk among others, that implements a novel approach for detecting mutational signatures.
In this vignette, we give an overview of the package by presenting some of its main functions.
Installing the RESOLVE R package
The RESOLVE package can be installed from Bioconductor as follow.
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("RESOLVE")Changelog
- 1.0.0 - Package released in Bioconductor 3.16.
- 1.2.0 - Major code refactoring.
- 1.10.0 - Major code update, implementing signatures-based clustering and associations to signatures exposures.
Using the RESOLVE R package
We now present some of the main features of the package. We notice that the package supports different types of mutational signatures such as: SBS (single base substitutions) and MNV (multi-nucleotide variant) (see Degasperi, Andrea, et al. “Substitution mutational signatures in whole-genome–sequenced cancers in the UK population.” Science 376.6591 (2022): abl9283), CX (chromosomal instability) (see Drews, Ruben M., et al. “A pan-cancer compendium of chromosomal instability.” Nature 606.7916 (2022): 976-983) and CN (copy number) signatures (see Steele, Christopher D., et al. “Signatures of copy number alterations in human cancer.” Nature 606.7916 (2022): 984-991). But, for the sake of this vignette, we present only results on the classical SBS signatures. We refer to the manual for details.
First, we show how to load example data and import them into a count matrix to perform the signatures analysis.
These data are a reduced version of the 560 breast tumors provided by Nik-Zainal, Serena, et al. (2016) comprising only 3 patients. We notice that these data are provided purely as an example, and, as they are a reduced and partial version of the original dataset, they should not be used to draw any biological conclusion.
We now import such data into a count matrix to perform the signatures discovery. To do so, we also need to specify the reference genome as a BSgenome object to be considered. This can be done as follows, where in the example we used hs37d5 as reference genome as provided as data object within the package.
library("BSgenome.Hsapiens.1000genomes.hs37d5")
imported_data = getSBSCounts(data = ssm560_reduced, reference = BSgenome.Hsapiens.1000genomes.hs37d5)Now, we present an example of visualization feature provided by the package, showing the counts for the first patient, i.e., PD10010a, in the following plot.
patientsSBSPlot(trinucleotides_counts=imported_data,samples="PD10010a")## Warning: `aes_string()` was deprecated in ggplot2 3.0.0.
## ℹ Please use tidy evaluation idioms with `aes()`.
## ℹ See also `vignette("ggplot2-in-packages")` for more information.
## ℹ The deprecated feature was likely used in the RESOLVE package.
## Please report the issue at <https://github.com/danro9685/RESOLVE/issues>.
## This warning is displayed once per session.
## Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
## generated.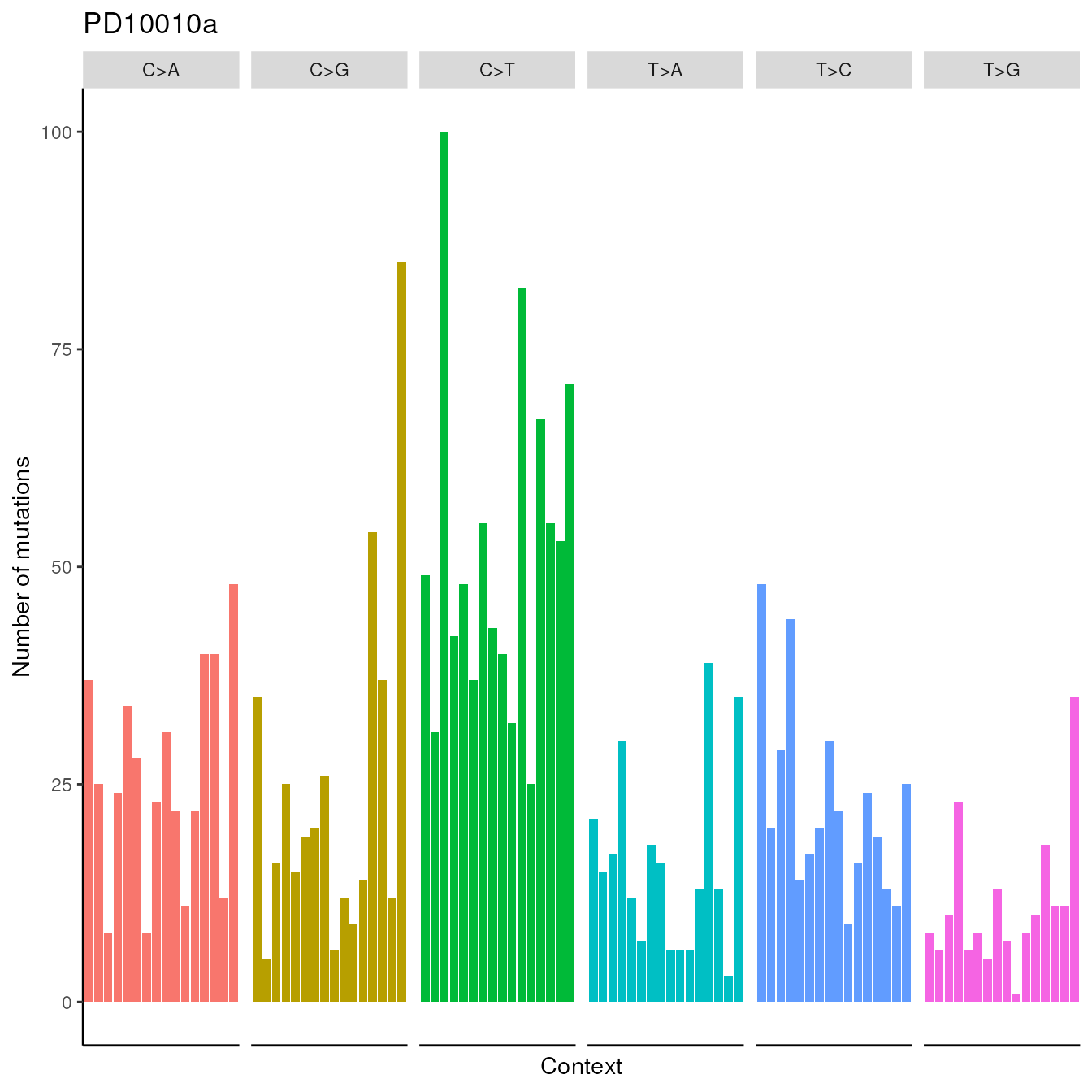
Visualization of the counts for patient PD10010a from the dataset published in Nik-Zainal, Serena, et al.
After the data are loaded, we can perform signatures de novo extraction. To do so, we need to define a range for the number of signatures (variable K) to be considered. We now show how to perform the inference on the dataset from Nik-Zainal, Serena, et al. (2016), whose counts are provided within the package.
data(background)
data(patients)
set.seed(12345)
res_denovo = signaturesDecomposition(x = patients,
K = 1:15,
background_signature = background,
nmf_runs = 100,
num_processes = 50)We notice that this function can be also used to perform de novo estimation for other types of mutational signatures, such as SBS, MNV, CX and CN.
Now that we have performed the de novo inferece, we need to decide the optimal number of signatures to be extracted from our dataset. To do so, we provide a procedure based on cross-validation.
set.seed(12345)
res_cv = signaturesCV(x = patients,
beta = res_denovo$beta,
cross_validation_repetitions = 100,
num_processes = 50)The computations for this task can be very time consuming, especially when many iterations of cross validations are performed (see manual) and a large set of configurations of the parameters are tested.
We refer to the manual for a detailed description of each parameter and to the RESOLVE manuscript for details on the method.
Signatures-based clustering and associations to signatures exposures
RESOLVE also allows researchers to perform downstream analyses once mutational signatures have been inferred. 1) It enables signatures-based clustering, using the k-medoids method to group patients with similar active mutational processes. Additionally, 2) it provides features to explore associations between signatures and somatic mutations, identifying links both from signatures to specific mutations and vice versa. Finally, 3) RESOLVE can examine the relationship between signature exposures and prognosis, providing insights into the clinical implications of different mutational processes. By integrating these analyses, RESOLVE enhances our understanding of the biological and clinical relevance of mutational signatures.
We next describe how to perform these three analyses with the RESOLVE R package.
Signatures-based clustering can be performed with the signaturesClustering functions, which takes as input exposures of signatures to patients. We suggest to provide normalized exposures.
data(sbs_assignments)
set.seed(12345)
norm_alpha = (sbs_assignments$alpha / rowSums(sbs_assignments$alpha))
sbs_clustering = signaturesClustering(alpha = norm_alpha, num_clusters = 1:3, num_processes = 1, verbose = FALSE)Associations of signatures exposures to somatic mutations in driver genes can be performed either with the associationAlterations function or the associationSignatures function via regularized regression. In the first case, mutations are the predictors of the regression and the signatures exposures the target varibles, which in the second case regularized logistic regression is performed with the mutations being the target varibles.
data(association_mutations)
set.seed(12345)
alterations = association_mutations$alterations
normalized_alpha = association_mutations$normalized_alpha
association_alterations = associationAlterations(alterations = alterations, signatures = normalized_alpha)
data(association_mutations)
set.seed(12345)
alterations = association_mutations$alterations
normalized_alpha = association_mutations$normalized_alpha
association_signatures = associationSignatures(alterations = alterations, signatures = normalized_alpha)Finally, signatures exposures can be associated to prognosis via regularized Cox regression with the associationPrognosis function. The results of this analysis are a set of coefficients measuring the impact of each signatures to prognosis, with positive values representing positive association with risk (bad prognosis) and negative values representing negative association with risk (better prognosis).
data(association_survival)
set.seed(12345)
clinical_data = association_survival$clinical_data
normalized_alpha = association_survival$normalized_alpha
prognosis_associations = associationPrognosis(clinical_data = clinical_data, signatures = normalized_alpha)Also in this case, we refer to the manual for a detailed description of each parameter and to the RESOLVE manuscript for details on the method.
Current R Session
## R Under development (unstable) (2026-04-05 r89793)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] BSgenome.Hsapiens.1000genomes.hs37d5_0.99.1
## [2] BSgenome_1.79.1
## [3] rtracklayer_1.71.3
## [4] BiocIO_1.21.0
## [5] Biostrings_2.79.5
## [6] XVector_0.51.0
## [7] GenomicRanges_1.63.1
## [8] Seqinfo_1.1.0
## [9] IRanges_2.45.0
## [10] S4Vectors_0.49.1
## [11] RESOLVE_1.14.0
## [12] Biobase_2.71.0
## [13] BiocGenerics_0.57.0
## [14] generics_0.1.4
## [15] BiocStyle_2.39.0
##
## loaded via a namespace (and not attached):
## [1] bitops_1.0-9 gridExtra_2.3
## [3] rlang_1.2.0 magrittr_2.0.5
## [5] gridBase_0.4-7 otel_0.2.0
## [7] matrixStats_1.5.0 compiler_4.7.0
## [9] ggalluvial_0.12.6 systemfonts_1.3.2
## [11] vctrs_0.7.2 reshape2_1.4.5
## [13] stringr_1.6.0 pkgconfig_2.0.3
## [15] shape_1.4.6.1 crayon_1.5.3
## [17] fastmap_1.2.0 labeling_0.4.3
## [19] Rsamtools_2.27.1 rmarkdown_2.31
## [21] pracma_2.4.6 UCSC.utils_1.7.1
## [23] ragg_1.5.2 xfun_0.57
## [25] glmnet_4.1-10 cachem_1.1.0
## [27] cigarillo_1.1.0 GenomeInfoDb_1.47.2
## [29] jsonlite_2.0.0 SnowballC_0.7.1
## [31] DelayedArray_0.37.1 BiocParallel_1.45.0
## [33] parallel_4.7.0 cluster_2.1.8.2
## [35] R6_2.6.1 bslib_0.10.0
## [37] stringi_1.8.7 RColorBrewer_1.1-3
## [39] jquerylib_0.1.4 Rcpp_1.1.1
## [41] bookdown_0.46 SummarizedExperiment_1.41.1
## [43] iterators_1.0.14 knitr_1.51
## [45] Matrix_1.7-5 splines_4.7.0
## [47] nnls_1.6 tidyselect_1.2.1
## [49] abind_1.4-8 yaml_2.3.12
## [51] doParallel_1.0.17 codetools_0.2-20
## [53] curl_7.0.0 lattice_0.22-9
## [55] tibble_3.3.1 plyr_1.8.9
## [57] withr_3.0.2 S7_0.2.1
## [59] evaluate_1.0.5 desc_1.4.3
## [61] survival_3.8-6 MutationalPatterns_3.21.1
## [63] pillar_1.11.1 lsa_0.73.4
## [65] BiocManager_1.30.27 rngtools_1.5.2
## [67] MatrixGenerics_1.23.0 foreach_1.5.2
## [69] RCurl_1.98-1.18 ggplot2_4.0.2
## [71] scales_1.4.0 NMF_0.28
## [73] RhpcBLASctl_0.23-42 glue_1.8.0
## [75] tools_4.7.0 data.table_1.18.2.1
## [77] GenomicAlignments_1.47.0 registry_0.5-1
## [79] fs_2.0.1 XML_3.99-0.23
## [81] grid_4.7.0 colorspace_2.1-2
## [83] restfulr_0.0.16 cli_3.6.5
## [85] textshaping_1.0.5 S4Arrays_1.11.1
## [87] dplyr_1.2.1 gtable_0.3.6
## [89] sass_0.4.10 digest_0.6.39
## [91] SparseArray_1.11.13 rjson_0.2.23
## [93] htmlwidgets_1.6.4 farver_2.1.2
## [95] htmltools_0.5.9 pkgdown_2.2.0.9000
## [97] lifecycle_1.0.5 httr_1.4.8