Using the package
Daniele Ramazzotti
Avantika Lal
Keli Liu
Luca De Sano
Robert Tibshirani
Arend Sidow
April 07, 2026
Source:vignettes/v2_using_the_package.rmd
v2_using_the_package.rmdWe now present the main features of the package. To start, we show how to load data and transform them to a count matrix to perform the signatures discovery; first we load some example data provided in the package.
The SparseSignatures package can be installed from Bioconductor as follow.
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("SparseSignature")## Loading required package: NMF## Loading required package: registry## Loading required package: rngtools## Loading required package: cluster## NMF - BioConductor layer [OK] | Shared memory capabilities [NO: bigmemory] | Cores 2/2## To enable shared memory capabilities, try: install.extras('
## NMF
## ')##
## Attaching package: 'NMF'## The following object is masked from 'package:generics':
##
## fit## sample chrom start end ref alt
## <char> <char> <int> <int> <char> <char>
## 1: PD10014a 1 186484577 186484577 A C
## 2: PD10014a 7 141761948 141761948 G A
## 3: PD10014a 7 71266228 71266228 C T
## 4: PD10014a 8 82304475 82304475 A T
## 5: PD10014a 3 191275626 191275626 T A
## 6: PD10014a 4 135265376 135265376 C TThese data are a reduced version with only 3 patients of the 560 breast tumors provided by Nik-Zainal, Serena, et al. (2016). We can transform such input data to a count matrix to perform the signatures discovery with the function import.counts.data. To do so, we also need to specify the reference genome as a BSgenome object and the format of the 96 nucleotides to be considered. This can be done as follows, where in the example we use hs37d5 as our reference genome.
library("BSgenome.Hsapiens.1000genomes.hs37d5")## Loading required package: BSgenome## Loading required package: S4Vectors## Loading required package: stats4##
## Attaching package: 'S4Vectors'## The following object is masked from 'package:NMF':
##
## nrun## The following object is masked from 'package:utils':
##
## findMatches## The following objects are masked from 'package:base':
##
## expand.grid, I, unname## Loading required package: IRanges## Loading required package: Seqinfo## Loading required package: GenomicRanges## Loading required package: Biostrings## Loading required package: XVector##
## Attaching package: 'Biostrings'## The following object is masked from 'package:base':
##
## strsplit## Loading required package: BiocIO## Loading required package: rtracklayer## context alt cat
## <char> <fctr> <char>
## 1: A:A C>A A[C>A]A
## 2: C:A C>A C[C>A]A
## 3: G:A C>A G[C>A]A
## 4: T:A C>A T[C>A]A
## 5: A:A C>G A[C>G]A
## 6: C:A C>G C[C>G]A
imported_data = import.trinucleotides.counts(data=ssm560_reduced,reference=bsg)
head(imported_data)## A[C>A]A A[C>A]C A[C>A]G A[C>A]T A[C>G]A A[C>G]C A[C>G]G A[C>G]T
## PD10010a 37 25 8 24 35 5 16 25
## PD10011a 103 59 16 73 113 54 31 102
## PD10014a 235 241 37 234 158 71 26 180
## A[C>T]A A[C>T]C A[C>T]G A[C>T]T A[T>A]A A[T>A]C A[T>A]G A[T>A]T
## PD10010a 49 31 100 42 21 15 17 30
## PD10011a 116 73 228 109 61 70 56 165
## PD10014a 229 89 178 186 105 90 126 174
## A[T>C]A A[T>C]C A[T>C]G A[T>C]T A[T>G]A A[T>G]C A[T>G]G A[T>G]T
## PD10010a 48 20 29 44 8 6 10 23
## PD10011a 184 116 113 169 77 41 73 105
## PD10014a 261 122 167 211 76 27 84 59
## C[C>A]A C[C>A]C C[C>A]G C[C>A]T C[C>G]A C[C>G]C C[C>G]G C[C>G]T
## PD10010a 34 28 8 23 15 19 20 26
## PD10011a 105 75 30 102 60 37 22 65
## PD10014a 244 238 35 243 107 105 40 144
## C[C>T]A C[C>T]C C[C>T]G C[C>T]T C[T>A]A C[T>A]C C[T>A]G C[T>A]T
## PD10010a 48 37 55 43 12 7 18 16
## PD10011a 71 52 108 103 116 80 89 103
## PD10014a 136 124 144 197 116 139 145 217
## C[T>C]A C[T>C]C C[T>C]G C[T>C]T C[T>G]A C[T>G]C C[T>G]G C[T>G]T
## PD10010a 14 17 20 30 6 8 5 13
## PD10011a 103 78 102 158 40 65 55 188
## PD10014a 103 144 112 129 47 54 70 107
## G[C>A]A G[C>A]C G[C>A]G G[C>A]T G[C>G]A G[C>G]C G[C>G]G G[C>G]T
## PD10010a 31 22 11 22 6 12 9 14
## PD10011a 78 50 14 55 55 66 13 87
## PD10014a 146 126 24 160 63 70 25 120
## G[C>T]A G[C>T]C G[C>T]G G[C>T]T G[T>A]A G[T>A]C G[T>A]G G[T>A]T
## PD10010a 40 32 82 25 6 6 6 13
## PD10011a 76 63 118 81 69 41 56 86
## PD10014a 141 99 180 163 62 66 83 126
## G[T>C]A G[T>C]C G[T>C]G G[T>C]T G[T>G]A G[T>G]C G[T>G]G G[T>G]T
## PD10010a 22 9 16 24 7 1 8 10
## PD10011a 96 62 82 93 56 46 35 99
## PD10014a 110 81 102 135 32 18 61 78
## T[C>A]A T[C>A]C T[C>A]G T[C>A]T T[C>G]A T[C>G]C T[C>G]G T[C>G]T
## PD10010a 40 40 12 48 54 37 12 85
## PD10011a 78 80 12 83 116 104 29 194
## PD10014a 202 191 17 253 198 159 33 325
## T[C>T]A T[C>T]C T[C>T]G T[C>T]T T[T>A]A T[T>A]C T[T>A]G T[T>A]T
## PD10010a 67 55 53 71 39 13 3 35
## PD10011a 119 94 78 126 121 43 64 91
## PD10014a 188 153 93 184 124 89 73 221
## T[T>C]A T[T>C]C T[T>C]G T[T>C]T T[T>G]A T[T>G]C T[T>G]G T[T>G]T
## PD10010a 19 13 11 25 18 11 11 35
## PD10011a 125 79 83 113 68 90 140 251
## PD10014a 143 118 75 148 71 54 76 160The function import.counts.data can also take a text file as input with the same format as the one shown above. Now, we show an example of a visualization feature provided by the package, and we show the counts for the first patient PD10010a in the following plot.
patients.plot(trinucleotides_counts=imported_data,samples="PD10010a")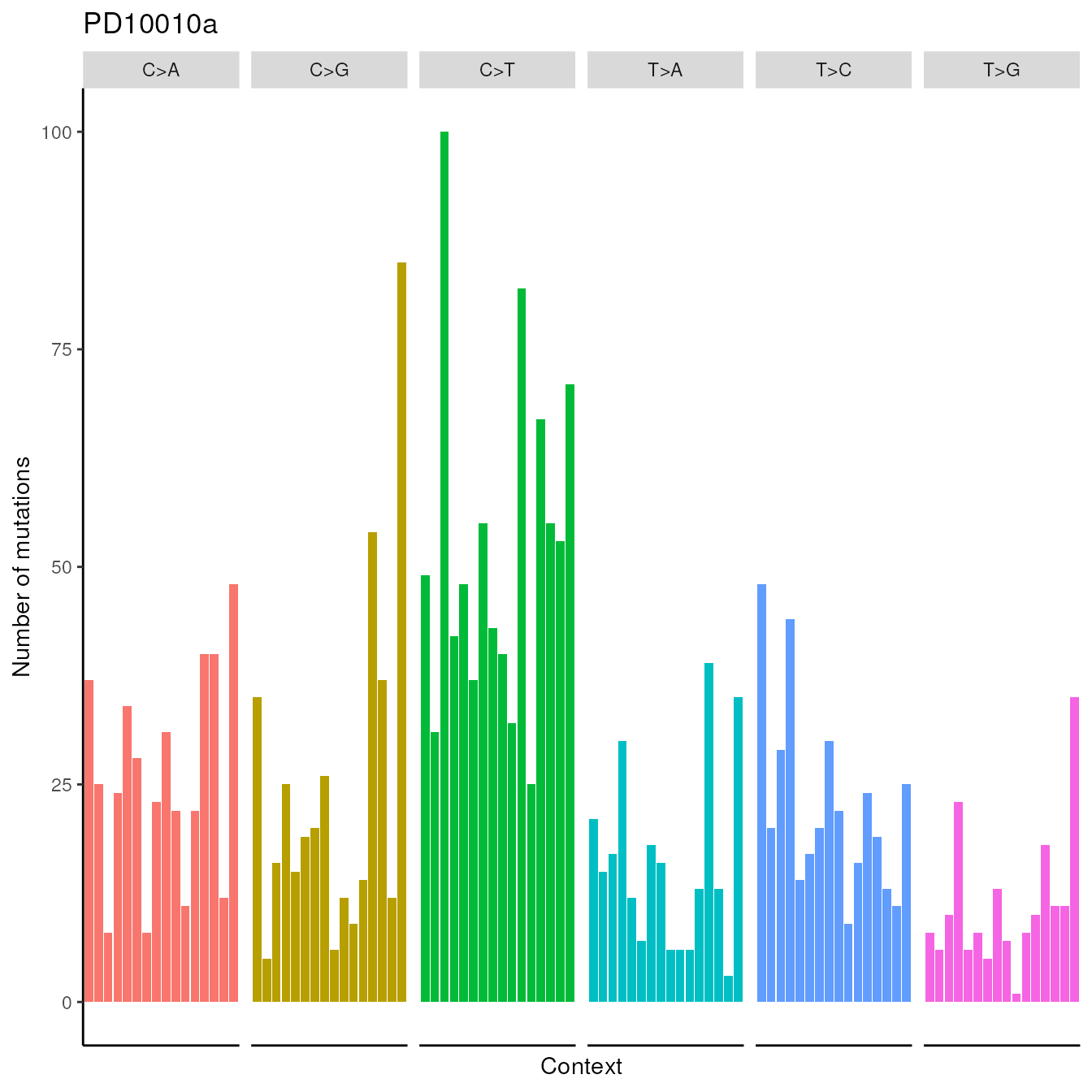
Visualization of the counts from patient PD10010a from the dataset published in Nik-Zainal, Serena, et al.
After the data are loaded, signatures can be discovered. To do so, we need to define a set of parameters on which to perform the estimation.
First of all, we need to specify the ranges for the number of signatures (variable K) and the LASSO penalty value (variable lambda rate) to be considered. The latter is more complicated to estimate, as it requires that the values in the range not to be too small in order to avoid dense signatures, but also should not be to high in order to still perform a good fit of the observed counts.
Besides these parameters, we also need to estimate the initial values of beta to be used during the estimation. We now show how to do this on the set of counts from 560 tumors provided in Nik-Zainal, Serena, et al. (2016).
## A[C>A]A A[C>A]C A[C>A]G A[C>A]T A[C>G]A A[C>G]C A[C>G]G A[C>G]T A[C>T]A
## PD8623a 24 23 4 20 10 19 2 11 43
## PD8618a 29 19 2 15 11 12 2 8 31
## PD6418a 23 29 4 26 12 9 1 12 39
## PD7214a 19 20 5 18 11 5 4 7 30
## PD4968a 59 64 5 34 25 16 1 18 81
## PD4954a 102 87 19 82 80 48 13 88 117
## A[C>T]C A[C>T]G A[C>T]T A[T>A]A A[T>A]C A[T>A]G A[T>A]T A[T>C]A A[T>C]C
## PD8623a 25 77 28 16 12 23 37 57 7
## PD8618a 17 91 24 10 10 8 18 50 23
## PD6418a 36 104 36 13 19 26 22 53 19
## PD7214a 22 65 21 12 18 17 18 41 12
## PD4968a 57 246 70 26 46 53 66 93 39
## PD4954a 53 125 79 64 48 37 52 97 41
## A[T>C]G A[T>C]T A[T>G]A A[T>G]C A[T>G]G A[T>G]T C[C>A]A C[C>A]C C[C>A]G
## PD8623a 30 42 12 6 8 16 32 21 6
## PD8618a 31 59 1 3 6 7 18 15 3
## PD6418a 32 57 7 4 6 8 24 19 2
## PD7214a 23 43 4 5 3 9 15 13 1
## PD4968a 47 85 17 6 7 16 45 27 10
## PD4954a 64 97 26 11 38 41 100 90 18
## C[C>A]T C[C>G]A C[C>G]C C[C>G]G C[C>G]T C[C>T]A C[C>T]C C[C>T]G C[C>T]T
## PD8623a 26 13 13 4 19 32 40 73 31
## PD8618a 14 4 9 4 3 21 33 61 30
## PD6418a 23 15 15 4 8 42 36 71 51
## PD7214a 10 7 5 2 12 31 32 48 40
## PD4968a 53 13 15 14 27 82 88 145 79
## PD4954a 83 77 48 22 65 90 64 84 99
## C[T>A]A C[T>A]C C[T>A]G C[T>A]T C[T>C]A C[T>C]C C[T>C]G C[T>C]T C[T>G]A
## PD8623a 10 10 10 11 14 15 15 23 3
## PD8618a 6 4 7 5 11 17 10 13 4
## PD6418a 6 13 9 14 19 8 13 14 6
## PD7214a 9 4 3 6 8 9 9 8 0
## PD4968a 13 25 20 36 22 24 29 37 7
## PD4954a 41 48 55 57 46 53 40 74 17
## C[T>G]C C[T>G]G C[T>G]T G[C>A]A G[C>A]C G[C>A]G G[C>A]T G[C>G]A G[C>G]C
## PD8623a 7 14 15 13 20 3 13 9 2
## PD8618a 4 6 5 17 13 9 14 2 10
## PD6418a 8 8 14 20 20 9 16 5 6
## PD7214a 7 8 12 24 7 2 8 6 6
## PD4968a 10 7 24 35 25 12 30 9 13
## PD4954a 19 37 42 53 67 13 42 40 28
## G[C>G]G G[C>G]T G[C>T]A G[C>T]C G[C>T]G G[C>T]T G[T>A]A G[T>A]C G[T>A]G
## PD8623a 1 6 33 24 61 29 3 11 6
## PD8618a 0 5 23 33 67 29 3 12 4
## PD6418a 3 5 35 39 94 34 7 12 9
## PD7214a 3 4 31 47 50 24 1 8 6
## PD4968a 1 11 68 62 190 65 8 21 14
## PD4954a 1 63 72 69 85 67 19 29 22
## G[T>A]T G[T>C]A G[T>C]C G[T>C]G G[T>C]T G[T>G]A G[T>G]C G[T>G]G G[T>G]T
## PD8623a 6 15 10 6 23 1 3 5 4
## PD8618a 5 17 10 8 23 0 1 1 0
## PD6418a 8 36 11 22 22 1 3 3 6
## PD7214a 8 26 12 8 18 1 3 2 2
## PD4968a 18 43 19 29 35 6 3 3 11
## PD4954a 49 61 37 34 54 12 7 32 36
## T[C>A]A T[C>A]C T[C>A]G T[C>A]T T[C>G]A T[C>G]C T[C>G]G T[C>G]T T[C>T]A
## PD8623a 34 24 8 31 22 20 1 32 119
## PD8618a 22 17 10 25 15 14 1 30 47
## PD6418a 34 23 5 35 9 12 2 24 43
## PD7214a 14 22 6 24 9 7 2 24 52
## PD4968a 79 57 9 87 64 27 8 120 464
## PD4954a 92 109 11 106 158 89 17 279 166
## T[C>T]C T[C>T]G T[C>T]T T[T>A]A T[T>A]C T[T>A]G T[T>A]T T[T>C]A T[T>C]C
## PD8623a 59 52 98 29 15 6 18 25 17
## PD8618a 26 37 37 20 4 3 13 21 12
## PD6418a 56 52 65 31 9 9 15 25 17
## PD7214a 38 41 62 14 8 7 16 19 14
## PD4968a 177 157 337 127 20 19 42 41 42
## PD4954a 114 48 150 62 44 27 71 58 38
## T[T>C]G T[T>C]T T[T>G]A T[T>G]C T[T>G]G T[T>G]T
## PD8623a 11 26 9 11 10 27
## PD8618a 12 16 4 3 6 11
## PD6418a 9 36 9 6 9 20
## PD7214a 13 22 4 10 8 19
## PD4968a 23 44 15 8 15 38
## PD4954a 30 57 40 29 37 62First, we can estimate the initial values of beta as follows.
starting_betas = startingBetaEstimation(x=patients,K=3:12,background_signature=background)Then, we also need to explore the search space of values for the LASSO penalty in order to make a good choice. To do so, we can use the function lambdaRangeBetaEvaluation to test different values to sparsify beta as follows. Notice that the package also provides the option to sparsify alpha and, in this case, we may use the function lambdaRangeAlphaEvaluation to explore the search space of values.
lambda_range = lambdaRangeBetaEvaluation(x=patients,K=10,beta=starting_betas[[8,1]],
lambda_values=c(0.05,0.10))As the executions of these functions can be very time-consuming, we also provide as examples together with the package a set of pre-computed results by the two functions startingBetaEstimation and lambdaRangeBetaEvaluation obtained with the commands above.
Now that we have evaluated all the required parameters, we need to decide which configuration of number of signatures and lambda value is the best. To do so, we rely on cross-validation.
cv = nmfLassoCV(x=patients,K=3:10)We notice that the computations for this task can be very time consuming, expecially when many iterations of cross validations are specified (see manual) and a large set of configurations of the parameters are tested. To speed up the execution, we suggest using the parallel execution options. Also, to reduce the memory requirements, we advise splitting the cross validation in different runs, e.g., if one wants to perform 100 iterations, we would suggest making 10 independent runs of 10 iterations each. Also in this case, we provide as examples together with the package a set of pre-computed results obtained with the above command and the following settings: K = 3:10, cross validation entries = 0.10, lambda values = c(0.05,0.10,0.15), number of iterations of cross-validation = 2.
data(cv_example)Finally, we can compute the signatures for the best configuration, i.e., K = 5.
beta = starting_betas_example[["5_signatures","Value"]]
res = nmfLasso(x = patients, K = 5, beta = beta, background_signature = background, seed = 12345)## Performing the discovery of the signatures by NMF with Lasso...
## Performing a total of 30 iterations...
## Progress 3.33333333333333%...
## Progress 6.66666666666667%...
## Progress 10%...
## Progress 13.3333333333333%...
## Progress 16.6666666666667%...
## Progress 20%...
## Progress 23.3333333333333%...
## Progress 26.6666666666667%...
## Progress 30%...
## Progress 33.3333333333333%...
## Progress 36.6666666666667%...
## Progress 40%...
## Progress 43.3333333333333%...
## Progress 46.6666666666667%...
## Progress 50%...
## Progress 53.3333333333333%...
## Progress 56.6666666666667%...
## Progress 60%...
## Progress 63.3333333333333%...
## Progress 66.6666666666667%...
## Progress 70%...
## Progress 73.3333333333333%...
## Progress 76.6666666666667%...
## Progress 80%...
## Progress 83.3333333333333%...
## Progress 86.6666666666667%...
## Progress 90%...
## Progress 93.3333333333333%...
## Progress 96.6666666666667%...
## Progress 100%...## Warning in nmfLassoDecomposition(x, beta, lambda_rate_alpha, lambda_rate_beta,
## : The likelihood is not increasing, you should try a lower value of lambda!
## Current settings: K = 6, lambda_rate_alpha = 0.05, lambda_rate_beta = 0.05...We conclude this vignette by plotting the discovered signatures.
data(nmf_LassoK_example)
signatures = nmf_LassoK_example$beta
signatures.plot(beta=signatures, xlabels=FALSE)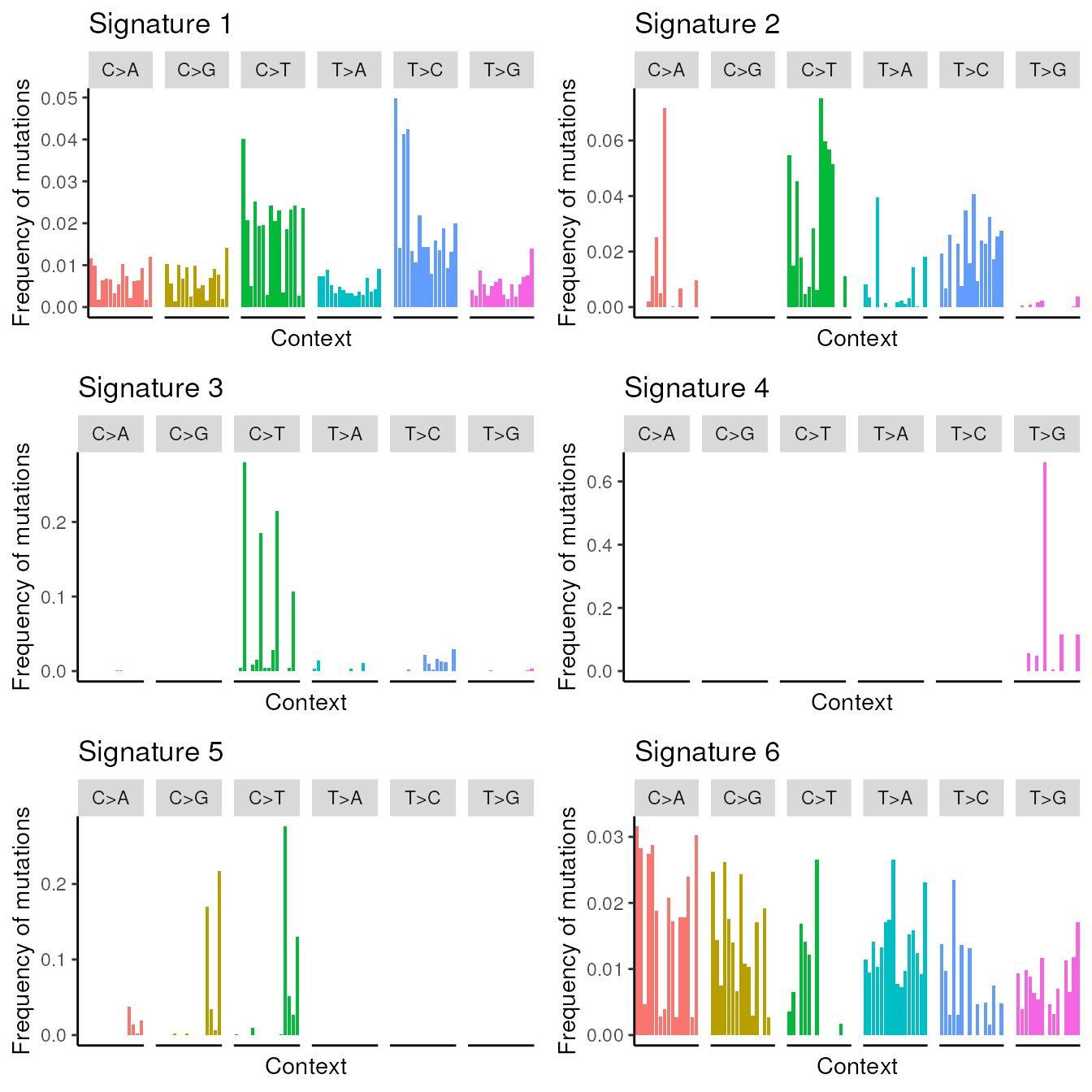
Visualization of the discovered signatures.